
Wenn wir ehrlich sind, wissen wir meist nicht genau, was wir einnehmen oder wie häufig eine Nebenwirkung auftreten kann, wenn wir unsere Kopfschmerztablette, das Asthmamittel oder die Blutdrucktablette schlucken. Wir verlassen uns darauf, dass die Medikamente ausgiebig getestet wurden und staatliche Behörden sie für unbedenklich erachten. Auf den ersten Blick scheint die staatliche Medikamentenzulassung nur Vorteile zu haben.
Doch die aufwendigen staatlich geforderten Tests und Zulassungsverfahren führen nicht nur zu höheren Kosten für das Medikament, sondern bergen zudem Kosten, die nicht auf den ersten Blick sichtbar sind: Menschen die von einem getesteten Medikament gesundheitlich profitieren könnten, erhalten es erst verspätet oder gar nicht.
Ein aktuelles Beispiel verdeutlicht dies. Forscher in den USA haben erfolgreich eine Therapie für die seltene Blutkrankheit Hämophilie B entwickelt, die bisher als unheilbar galt. Die Therapie würde für die Betroffenen eine enorme Verbesserung ihrer Lebensumstände bedeuten. Doch Professor Oldenburg vom Universitätsklinikum Bonn dämpft die Hoffnung auf eine schnelle Verfügbarkeit:
„Dennoch wird es sicher noch drei bis fünf Jahre dauern, bis diese Therapie den meisten Hämophilie B-Patienten zur Verfügung steht.“
Vom Labor in die Apotheke
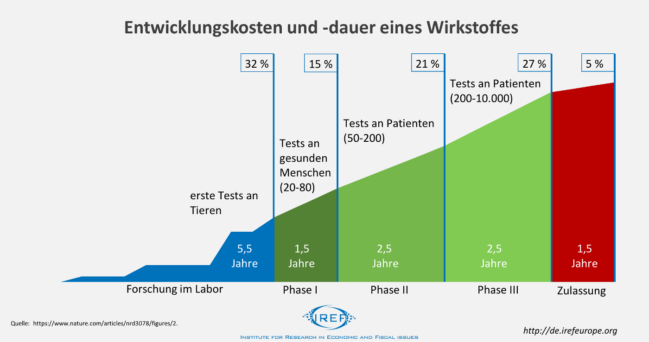
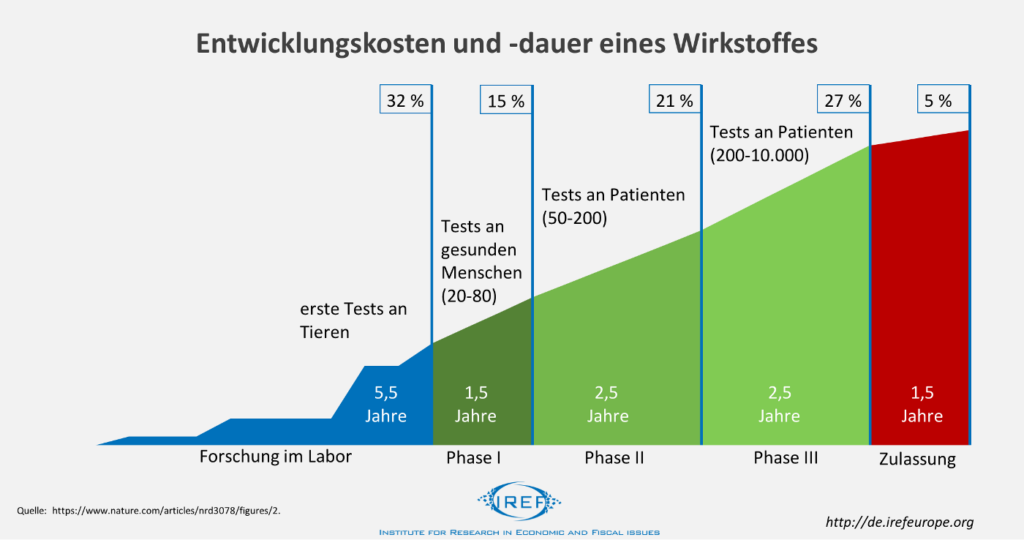
Durchschnittlich dauert die Entwicklung und Zulassung eines neuen Wirkstoffes 13,5 Jahre. In den ersten 5,5 Jahren wird der Wirkstoff in Labors untersucht und die Wirksamkeit sowie mögliche schädliche Wirkungen an Tieren getestet. Anschließend folgen in drei Phasen 6,5 Jahre klinische Tests an Menschen. In der ersten Phase werden an gesunden Menschen Tests auf Verträglichkeit durchgeführt. In der zweiten Phase wird der Wirkstoff an wenigen Patienten getestet, um in der dritten Phase an mehreren tausend Patienten erprobt zu werden. Absolviert der Wirkstoff diese Phasen erfolgreich, können die Entwickler die Zulassung beantragen und reichen dafür die Ergebnisse der vorherigen Tests bei den Zulassungsbehörden ein.
Die Zulassung: Sichtbare und nicht sichtbare Fehler
Der letzte Schritt, die Zulassung, ist eine kritische Phase. Hier entscheidet sich, ob sich die vorangegangenen Mühen auszahlen. Irren ist menschlich. Auch Zulassungsbehörden können irren. Dabei können sie zwei Arten von Fehlern begehen. Erstens können sie ein Medikament zulassen, das schädlich ist. Zweitens können sie einem Medikament die Zulassung verwehren, das unbedenklich ist und so Menschen helfen könnte. Beide Fehler führen dazu, dass die Gesundheit von Menschen gefährdet wird. In manchen Fällen hängen von beiden Arten von Fehlern Leben ab.
Eine fatale Asymmetrie
Der Fehler eines zugelassenen schädlichen Präparats wird automatisch korrigiert. Sollte ein Medikament sich als schädlich herausstellen, wird es vom Markt genommen. Der zweite Fehler wird hingegen nicht automatisch korrigiert. Wird ein Medikament nicht zugelassen, wird zu einem späteren Zeitpunkt auch nicht entdeckt, dass es Menschen hätte helfen können.
Schon 1972 kam der Ökonom Sam Peltzman in einer Studie zu dem Schluss, dass zu späte Zulassungen oder nicht erteilte Zulassungen von brauchbaren Medikamenten mehr geschadet haben als die Verhinderung unbrauchbarer Medikamente. Seitdem gab es einige Bemühungen die Zulassungen zu beschleunigen. So wurde beispielsweise 1992 in den USA ein entsprechendes Gesetz erlassen. Eine Studie aus dem Jahr 2008 kommt zu dem Schluss, dass die daraufhin schneller erteilten Zulassungen bis zu 310.000 Menschenleben gerettet haben.
Dieser Befund deutet zum einen darauf hin, dass durch eine weitere Beschleunigung möglicherweise zukünftig abermals mehr Leben gerettet werden könnten. Zum anderen führt er vor Augen, dass das durch nicht erfolgte Zulassungen verursachte Leid potentiell beträchtlich ist. Denn die Zulassungsbehörden sehen sich einer außergewöhnlichen Anreizstruktur ausgesetzt.
Bedenkliche Anreize für Zulassungsbehörden
Schädliche zugelassene Medikamente führen zu sichtbaren Opfern und entsprechende Reaktionen der Öffentlichkeit. Eines der wohl tragischsten Beispiele ist der Wirkstoff Thalidomid und der damit verbundene Contergan-Skandal. Die staatlichen Zulassungsstellen haben einen starken Anreiz, diese Art von Fehlern zu vermeiden.
Opfer des zweiten Fehlers bleiben dagegen im Verborgenen. Opfer des Fehlers, ein Medikament nicht zuzulassen, das hätte zugelassen werden sollen, bleiben unsichtbar. Der Anreiz der Zulassungsbehörden, derartige Fehler zu vermeiden, fällt deutlich schwächer aus. Reaktionen der Öffentlichkeit müssen sie in diesen Fällen kaum fürchten.
Medikamente sehr ausgiebig in langwierigen Verfahren zu testen und im Zweifel nicht zuzulassen, hilft den Zulassungsbehörden folglich bei der Vermeidung sichtbarer Fehler und trägt zu mehr als der optimalen Anzahl unsichtbarer Fehler bei.
Ein ehemaliger Mitarbeiter der amerikanischen
Medikamentenzulassungsbehörde beschreibt einen Fall, in dem die Behörde davon überzeugt war, dass das betreffende Medikament effektiv und unbedenklich sei. Sein Vorgesetzter weigerte sich dennoch die Zulassung zu erteilen – mit der folgenden Begründung:
„Wenn irgendwas schief geht (…) denken Sie daran, wie übel es aussieht, wenn wir das Medikament so schnell genehmigt haben.“
Schnellere Zulassungen in den USA
Seit 1997 gibt es in den USA eine „Fast Track–Zulassung“ von Medikamenten für besonders schwerwiegende und lebensbedrohliche Krankheiten. Das Konzept setzt bei den der Zulassung vorgelagerten klinischen Tests an, die gut die Hälfte der gesamten Entwicklungsdauer vereinnahmen – hier ist offensichtlich das größte Zeitersparnispotential zu finden. Die Medikamente werden schrittweise für verschiedene Patientengruppen zugelassen, noch bevor größer angelegte Feldstudien durchgeführt werden. Der Kreis der Patienten und die Zulassung werden ausgeweitet, wenn Erfahrungen über das Risiko-Nutzen-Verhältnis mit den ersten Patientengruppen gesammelt wurden.
Auch Europa sammelt Erfahrung
Die Europäische Arzneimittel-Agentur hat im Jahre 2005 sogenannte „Conditional Approval“ für Medikamente eingeführt. Diese sollen ähnlich wie in den USA ermöglichen, dass Patienten, die unter besonders schwerwiegenden Krankheiten leiden, schneller Zugang zu neuen Medikamenten bekommen. Dabei werden Medikamente unter der Auflage, dass Phase III nachgeholt wird, bereits nach Phase II zugelassen. Dies ermöglicht einen bis zu 2,5 Jahre schnelleren Zugang zum entsprechenden Medikament.
Schnelle Zulassungen genauso sicher wie konventionelle
In einer Studie aus dem Jahr 2011 wurde untersucht, ob Medikamente, die in der EU in einem Schnellzulassungsverfahren zugelassen wurden, ein höheres Risiko aufweisen als konventionell zugelassene Medikamente. Dabei wurden nicht häufiger neue Risiken nach der Zulassung erkannt, als bei konventionell zugelassenen Medikamenten, die die Phasen I bis III komplett durchliefen.
Gegenseitig Zulassungen anerkennen
Ein erster Schritt, Medikamente schneller und günstiger für Patienten verfügbar zu machen, wäre die gegenseitige Anerkennung der Zulassung von Medikamenten in den USA und Europa. In der Regel werden die gleichen Unterlagen für die jeweilige Zulassung eingereicht. Warum sollte ein Medikament, das für EU Bürger zugelassen wurde, für Amerikaner schädlich sein?
Schnellere Zulassungen allein genügen nicht
Doch schnellere Zulassungen und ihre gegenseitige Anerkennung allein genügen nicht. Sie führen nicht dazu, dass die Zulassungsbehörden einen schwächeren Anreiz haben, unbedenkliche Medikamente nicht zuzulassen. Es stellt sich daher die grundsätzliche Frage, ob eine zwingende staatliche Zulassung nötig ist.
Unkonventioneller Einsatz bereits beschränkt möglich
Schon jetzt zeigen Ärzte und Patienten, dass sie bereit sind, Medikamente zu verschreiben und zu konsumieren, die nicht spezifisch getestet oder von den Behörden für diese Anwendungen zugelassen sind. Der Off-Label-Use, also die Nutzung von Medikamenten außerhalb des von den Behörden zugelassen Bereichs, ist vor allem bei Kindern weit verbreitet. So waren 2002 in Deutschland etwa 57 % aller verordneten Medikamente für gesetzlich krankenversicherte Neugeborene nicht für diese zugelassen.
Zudem ist es bereits möglich, nicht zugelassenen Medikamente im Rahmen der Härtefall-Verordnung zu erhalten. Allerdings gibt es nur sehr wenige Ausnahmen. So sind derzeit nur sieben Medikamente beim Bundesinstitut für Arzneimittel und Medizinprodukte und drei weitere beim Paul Ehrlich Institut gelistet, die für Härtefälle verfügbar sind. Ferner sind nicht zugelassene Medikamente im Rahmen individueller Heilversuche erhältlich. Allerdings dürfen diese Medikamente nicht beworben werden. Der Patient oder der behandelnde Arzt muss also auf anderen Wegen auf den potentiell hilfreichen Wirkstoff aufmerksam werden.
Die Nutzung von nicht zugelassenen Medikamenten und der Einsatz außerhalb des zugelassenen Bereichs zeigen, dass Patienten und Ärzte in der Lage und willens sind, auch ohne staatliche Zulassung Risiko und Nutzen abzuwägen. Nicht bzw. noch nicht zugelassene Medikamente sollten Patienten leichter zugänglich gemacht werden als derzeit.
Freiwillige staatliche Prüfung
Natürlich gibt es Menschen, die ausschließlich auf die staatliche Zulassung vertrauen. Eine freiwillige staatliche Prüfung von Medikamenten, wodurch die Vermarktung auch noch nicht zugelassener Medikamenten möglich würde, wäre eine für beide Patientengruppen attraktive Lösung. Die Medikamente, die nicht dieser freiwilligen Prüfung unterzogen werden, könnten mit einem entsprechenden Hinweis versehen werden.
Medikamente schneller und umfangreicher verfügbar machen
Unsichtbare Opfer, die zu einem hilfreichen Medikament keinen Zugang haben, sollten nicht unberücksichtigt bleiben. Deshalb sollten in einem ersten Schritt die Zulassungsverfahren deutlich beschleunigt und die Zulassungen durch Stellen anderer Staaten anerkannt werden. Zudem sollte den bedenklichen Anreizen der Zulassungsbehörden durch freiwillige staatliche Prüfungen begegnet werden. Kosten könnten so reduziert und die Gesundheit vieler Menschen verbessert werden.
Erschienen bei: IREF.